

Sindromul Von Hippel Lindau reprezinta o forma de cancer familial, care se manifesta prin:
- angioamatoza sau hemangioblastoza retiniana, cerebeloasa si viscerala: pulmonara, hepatica, pancreatica, suprarenala;
- afectiuni oculare: cataracta, nistagmus, cecitate, glaucom, anomalii ale vaselor retiniene;
- simptomatologie neurologica: ataxie, diplegie/paraplegie/tetraplegie, hidrocefalie, agenezie, tulburari de vorbire, hidrocefalie;
- patologie oncologica: cancer renal cu celule clare, cancer pancreatic, hipernefrom, feocromocitom, paragangliom, adenocarcinom al ampulei Vater, adenom chistic al ligamentului larg si al epididimului, tumora sacului endolimfatic;
- boala polichistica renala, pancreatica si genitala;
- hipertensiune arteriala;
- policitemie.
Exista doua tipuri de sindrom Von Hippel Lindau:
- VHL tip 1: carcinom renal asociat cu hemangioblastom;
- VHL tip 2A: feocromocitom asociat cu hemangioblastom;
- VHL tip 2B: feocromocitom asociat cu carcinomul renal;
- VHL tip 2C: feocromocitom izolat.
Diagnosticul de sindrom Von Hippel Lindau se stabileste pe baza asocierii a 6 leziuni majore:
- Hemangioblastom de retina;
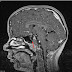
- Hemangioblastom cerebral (situat in special la nivelul cerebelului sau a maduvei spinarii);
- Feocromocitom;
- Carcinom renal cu celule clare sau boala polichistica renala;
- Tumori neuroendocrine ale pancreasului sau chiste pancreatice;
- Tumori ale sacului endolimfatic.
Pentru identificarea sindromului VLH se efectueaza:
- dozarea metanefrinelor urinare si efectuarea scintigrafiei cu metaiodo-benzy guanidina (MIBG) - in cazul suspicionarii unui feocromocitom;
- Ecografia si/sau CT-ul abdominal pentru identificarea tumorilor pancreatice, renale si suprarenale;
- Endoscopia digestiva pentru identificarea tumorilor endocrine pancreatice;
- RMN-ul pentru evidentierea hemangiomelor sistemului nervos, feocromocitoamelor sau a tumorilor de sac endolimfatic;
- Examenul fund de ochi sau angiografia pentru identificarea hemangioblastoamelor retiniene.
Tratamentul sindromului VHL consta in:
- chirurgie pentru formatiunile tumorale;
- crioterapie si fotocoagulare pentru leziunile retiniene;
- tratament medicamentos cu VEGF (blocanti ai factorului de crestere al endoteliului vascular) in cazul formatiunilor tumorale inoperabile.
sursa foto: http://www.mdanderson.org/patient-and-cancer-information/cancer-information/cancer-types/von-hippel-lindau-disease/vhl-full.jpg









Niciun comentariu:
Trimiteți un comentariu