

Afectiunea se datoreaza mutatiei genei NF-1 (implicata in sinteza neurofibrominei), localizata pe bratul lung al cromozomului 17 (17q11.2).
Semnele clinice specifice afectiunii sunt reprezentate de neurofibroame, pete cafe-au-lait, noduli Lisch, efelide axilare sau inghinale si tumori intracraniene.
Neurofibroamele pot fi cutanate (avand aspectul unor mici noduli sesili sau pediculati, de consistenta moala, in general nedurerosi), subcutanate sau plexiforme (localizarea cea mai frecventa fiind la nivelul plexului brahial, nervilor intercostali sau crurali). Leziunile sunt mai rare in perioada copilariei, insa se inmultesc odata cu inaintarea in varsta. S-a constatat ca perioada pubertara si cea gestationala determina aparitia acestor formatiuni si dezvoltarea rapida a celor preexistente.
 Petele cafe-au-lait apar in primii ani de viata crescand in dimensiuni si numar in perioada copilariei. In general sunt respectate palmele, plantele si scalpul. Alte semne specifice neurofibromatozei sunt pistruii (efelidele) cu distributie axilara sau inghinala.
Petele cafe-au-lait apar in primii ani de viata crescand in dimensiuni si numar in perioada copilariei. In general sunt respectate palmele, plantele si scalpul. Alte semne specifice neurofibromatozei sunt pistruii (efelidele) cu distributie axilara sau inghinala.
 Nodulii Lisch sunt hamartoame iriene melanocitare observate uneori cu ochiul liber sau la examinarea cu lampa cu fanta sub forma unor mici pete hiperpigmentate. Aceste leziuni oculare apar in 25% din cazuri in perioada copilariei si in peste 90% din cazuri la persoanele de varsta inaintata.
Nodulii Lisch sunt hamartoame iriene melanocitare observate uneori cu ochiul liber sau la examinarea cu lampa cu fanta sub forma unor mici pete hiperpigmentate. Aceste leziuni oculare apar in 25% din cazuri in perioada copilariei si in peste 90% din cazuri la persoanele de varsta inaintata.
Alte manifesari oculare aparute in cadrul neurofibromatozei sunt glaucomul congenital si gliomul optic. In cazuri extrem de rare (2-4%) poate aparea neurofibromul plexiform al pleoapei superioare (caracterizat prin ptoza palpebrala, hipertrofie hemifaciala homolaterala, hidrocefalie, buftalmie, cecitate, anomalii osoase ale viscerocraniului si ale bazei craniului) sau neurofibromul primitiv al orbitei (caracterizat prin exoftalmie asociata cu anomalii ale orbitei si ale boltii palatine).
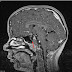
Tumorile cerebrale intalnite in cadrul neurofibromatozei sunt reprezentate de glioame (localizate la nivelul nervilor optici, chiasmei optice etc.), hamartoame, schwanoame, astrocitoame si meningioame.
Manifestarile osoase specifice bolii sunt reprezentate de cifoscolioza, pseudartroza oaselor lungi si displazia aripilor sfenoidului.
Alte manifestari care se pot asocia bolii sunt: macrocrania, hidrocefalia, retardul intelectual, epilepsia, talia mica, pubertatea precoce sau intarziata. Afectiunile maligne cele mai frecvent implicate in cadrul bolii sunt: leucemia mieloida cronica si neurofibrosarcomul.
In cazul in care neurofibromatoza se asociaza cu feocromocitomul se poate lua in discutie existenta sindromului MEN2B.
Diagnosticului de neurofibromatoza tip 1 se stabileste pe baza pozitivarii a cel putin doua din urmatoarele 7 criterii:
Tratamentul consta in corectie chirurgicala (in cazul decelarii formatiunilor tumorale), tratament ortopedic (pentru anomaliile scheletale) si tratament specific (pentru cefalee, convulsii etc).
sursa foto:
http://biomedicalephemera.tumblr.com/post/13786497416/michel-bur-case-study-2-of-dr-von
http://www.actasdermo.org/en/rasopathies-developmental-disorders-that-predispose/articulo/S1578219011000163/
http://www.milesresearch.com/main/eyesigns.asp


Alte manifesari oculare aparute in cadrul neurofibromatozei sunt glaucomul congenital si gliomul optic. In cazuri extrem de rare (2-4%) poate aparea neurofibromul plexiform al pleoapei superioare (caracterizat prin ptoza palpebrala, hipertrofie hemifaciala homolaterala, hidrocefalie, buftalmie, cecitate, anomalii osoase ale viscerocraniului si ale bazei craniului) sau neurofibromul primitiv al orbitei (caracterizat prin exoftalmie asociata cu anomalii ale orbitei si ale boltii palatine).
Tumorile cerebrale intalnite in cadrul neurofibromatozei sunt reprezentate de glioame (localizate la nivelul nervilor optici, chiasmei optice etc.), hamartoame, schwanoame, astrocitoame si meningioame.
Manifestarile osoase specifice bolii sunt reprezentate de cifoscolioza, pseudartroza oaselor lungi si displazia aripilor sfenoidului.
Alte manifestari care se pot asocia bolii sunt: macrocrania, hidrocefalia, retardul intelectual, epilepsia, talia mica, pubertatea precoce sau intarziata. Afectiunile maligne cele mai frecvent implicate in cadrul bolii sunt: leucemia mieloida cronica si neurofibrosarcomul.
In cazul in care neurofibromatoza se asociaza cu feocromocitomul se poate lua in discutie existenta sindromului MEN2B.
Diagnosticului de neurofibromatoza tip 1 se stabileste pe baza pozitivarii a cel putin doua din urmatoarele 7 criterii:
- 6 pete "café au lait” (> 0,5 cm inainte de pubertate; > 1,5 cm după pubertate);
- 2 neurofibroame de orice tip sau un neurofibrom plexiform;
- pistrui axilari sau inghinali;
- glioame optice;
- 2 noduli Lisch;
- leziuni osoase (displazia aripilor osului sfenoid, pseudartoza oaselor lungi, scolioza);
- o ruda de gradul I cu neurofibromatoza tip 1 ce intruneste criteriile de mai sus.
Diagnosticul diferential al bolii se face cu: neurofibromatoza tip 2, sindromul Mccune-Albright, scleroza tuberoasa si sindromul Legius.
Tratamentul consta in corectie chirurgicala (in cazul decelarii formatiunilor tumorale), tratament ortopedic (pentru anomaliile scheletale) si tratament specific (pentru cefalee, convulsii etc).
sursa foto:
http://biomedicalephemera.tumblr.com/post/13786497416/michel-bur-case-study-2-of-dr-von
http://www.actasdermo.org/en/rasopathies-developmental-disorders-that-predispose/articulo/S1578219011000163/
http://www.milesresearch.com/main/eyesigns.asp









Niciun comentariu:
Trimiteți un comentariu